近日,药物化学权威期刊European Journal of Medicinal Chemistry(IF=7.088)发表了我校制药工程学院侯云雷/宫平课题组在新型PLK1抑制剂研究领域的最新成果,文章题目为“Design,synthesis,and biological evaluation of novel dihydropteridone derivatives possessing oxadiazoles moiety as potent inhibitors of PLK1”。
Polo样激酶(Polo-like kinases,PLKs)是一个广泛分布于真核细胞中的丝氨酸/苏氨酸(Ser/Thr)蛋白激酶家族,在细胞周期的几个阶段发挥重要作用。其中PLK1在细胞分裂、DNA复制/修复、DNA检查点调节和多种有丝分裂过程中表现出重要功能。作为原癌基因,PLK1在包括黑色素瘤、胶质母细胞瘤、乳腺癌、结肠癌、肺癌和胃癌等多种癌症中的表达水平升高,是治疗癌症的潜在药物靶点。
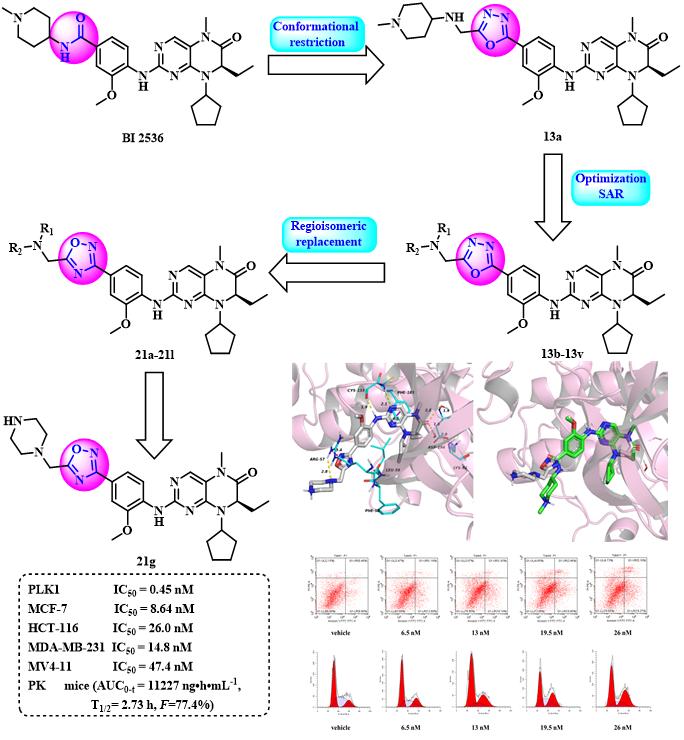
课题组采用基于结构的药物设计策略,以BI2536为先导化合物,运用构象限制和生物电子等排药物设计策略,得到了一系列含噁二唑片段的二氢蝶啶酮类衍生物,其中化合物21g表现出显著的PLK1抑制能力,其IC50值为0.45nM,并且对多种肿瘤细胞系具有显著的抗增殖活性(MCF-7IC50=8.64nM,HCT-116IC50=26.0nM,MDA-MB-231IC50=14.8nM和MV4-11IC50=47.4nM)。此外,化合物21g在小鼠中表现出比BI2536更好的药代动力学特性(AUC0-t=111227ng·h·mL-1vs556ng·h·mL-1),口服生物利用度为77.4%,并且在Balb/c小鼠急性毒性试验(20mg/kg)中未观察到明显的毒性。化合物21g可使HCT-116细胞阻滞在G2期,并以剂量依赖的方式诱导细胞凋亡。
我校2020级药物化学博士研究生李志威为文章第一作者,制药工程学院宫平教授、侯云雷副教授为共同通讯作者。
文章链接:https://www.sciencedirect.com/science/article/pii/S0223523423002088
文章DOI:https://doi.org/10.1016/j.ejmech.2023.115242