近日,化学领域知名期刊Angewandte Chemie International Edition《德国应用化学》(IF=16.9,Q1)在线发表了我校客座教授刘宏伟团队的最新研究成果“Iizuchalasin A: A Marine Fungal Metabolite With a Cage-Like Structure That Binds TarG to Inhibit Wall Teichoic Acid Biosynthesis by Multidrug-Resistant S. aureus”,报道了一种源自海洋真菌的全新抗生素-Iizuchalasin A,为对抗多药耐药"超级细菌"提供了新的先导分子。
全球抗生素耐药性问题日益严峻,多重耐药金黄色葡萄球菌(包括MRSA和MDRSA)等"超级细菌"对临床常用抗生素产生了极强的耐药性,严重威胁公共卫生安全。大多数现有抗生素的作用靶点(如核糖体、细胞壁合成酶等),细菌早已“熟记于心”,并发展出多种逃避策略。因此,如何另辟蹊径开发结构新颖、作用机制独特的新型抗生素已成为抗感染药物研究的关键前沿。
在药物发现的版图上,大分子量天然产物曾因其分离与结构解析的艰巨性,如同笼罩在迷雾中的神秘大陆,资源丰饶却难以开拓。然而,近年来从“不可培养”的土壤微生物中成功捕获了肽类抗生素 Teixobactin;通过对昆虫共生菌的挖掘,发现了能精准靶向革兰氏阴性菌外膜复合体的 Darobactin。这些复杂大分子的成功“破译”,不仅验证了大分子成药的巨大潜力,更为抗击耐药菌指明了新的战略方向。在此背景下,本研究直面全球耐药性挑战,聚焦于海洋真菌所合成的独特化学物质,致力于从中发现能有效制服多重耐药金黄色葡萄球菌的全新武器。
该化合物由中国科学院微生物研究所与沈阳药科大学团队合作通过质谱代谢组定向锁定分子量大于1000的真菌天然产物,从海洋真菌 Aspergillus iizukae 中分离获得,属于罕见的merocytochalasan类天然产物。其最引人注目的特征是高度对称的笼状“蝴蝶样”骨架——通过X射线单晶衍射解析,科学家发现它由两个相同的aspochalasin-epicoccine单元通过多点共价连接,形成包含17个环和16个手性中心的复杂三维结构。
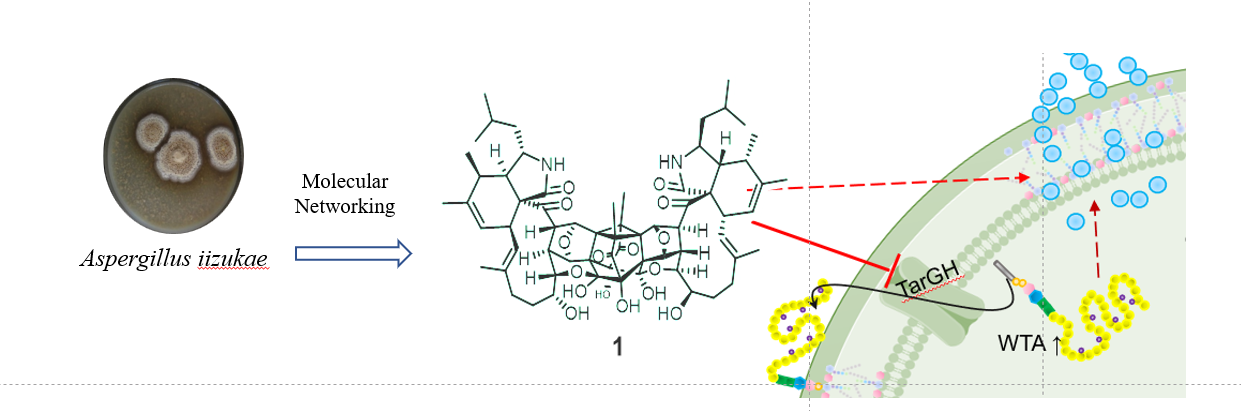
Iizuchalasin A对多重耐药金黄色葡萄球菌(包括MRSA和MDRSA)表现出较强抗菌活性,不仅能抑制细菌生长,还可促进囊泡的释放、显著降低其黏附能力与毒力因子表达。在小鼠皮肤感染模型中,局部给药即可大幅减少细菌负荷;在系统性感染模型中,显著提高小鼠存活率。更重要的是,其诱导耐药突变的能力低,且所获耐药突变株丧失毒力和致病性。
壁磷壁酸是革兰氏阳性菌细胞壁上的关键成分,对细菌的存活、分裂和致病都至关重要。机制研究表明,Iizuchalasin A特异性靶向革兰氏阳性菌细胞壁磷壁酸(WTA)转运蛋白TarGH-细胞壁关键组分磷壁酸合成的关键酶。该化合物结合TarG并抑制其ATP酶活性,从而阻断WTA前体外翻,破坏细胞壁完整性。这一作用模式虽与已知TarG抑制剂targocil类似,但突变谱和结合特性提示其具有独特的结合位点与作用方式。由于现有临床抗生素极少针对这一靶点,这意味着细菌对它几乎没有预存的耐药性,为临床治疗赢得了宝贵的时间窗口。
Iizuchalasin A的成功发现为抗耐药菌抗生素研制提供了独特的先导分子。作为分子量超1100 Da的大分子天然产物,它不仅展现出优异的体内疗效和低耐药风险,更以其新颖的“蝴蝶样”笼状结构开辟了靶向WTA通路的新化学空间。同时,也为天然产物化学研究向大分子化合物边界拓展提供了借鉴。研究团队认为,通过半合成修饰或结构简化,该分子有望发展为新一代抗MRSA候选药物,应对日益严峻的全球耐药危机。
我校2021级博士研究生张芮、天津科技大学陈保送副教授和我校王海峰副教授为论文的共同第一作者,刘宏伟研究员和微生物研究所代焕琴副研究员为本文的通讯作者,沈阳药科大学为第一通讯单位。
文章链接:https://onlinelibrary.wiley.com/doi/10.1002/anie.202523303